

Optimized Method for Robust Transcriptome Profiling of Minute Tissues Using Laser Capture Microdissection and Low-Input RNA-Seq
Shannon Farris, Yu Wang, James M. Ward, Serena M. Dudek.
Frontiers in Molecular Neuroscience (2017)
DOI: https://doi.org/10.3389/fnmol.2017.00185
PMID: 28659759
Publication
Abstract
Obtaining high quality RNA from complex biological tissues, such as the brain, is needed for establishing high-fidelity cell-type specific transcriptomes. Although combining genetic labeling techniques with laser capture microdissection (LCM) is generally sufficient, concerns over RNA degradation and limited yields call into question results of many sequencing studies. Here we set out to address both of these issues by: (1) developing a fluorescence-assisted LCM protocol that yields high quality RNA from fresh-frozen tissues; and (2) determining a suitable RNA-Seq library generation method for limited amounts of RNA (1-5 ng total RNA). The latter focused on comparing commercially available kits able to produce libraries of sufficient concentration and complexity while limiting PCR amplification biases. We find that high quality RNA (RNA integrity number, RIN, >9) of sufficient concentration can be isolated from laser-captured material from thinly-sectioned tissues when digestion time and temperature are minimized. Furthermore, we found that library generation approaches that retain ribosomal RNA (rRNA) through cDNA library generation required fewer cycles of PCR, minimizing bias in the resulting libraries. Lastly, end stage depletion of rRNA prior to sequencing enriches for target RNAs, thereby increasing read depth and level of gene detection while decreasing sequencing costs. Here we describe our protocol for generating robust RNA-Seq libraries from laser-captured tissue and demonstrate that with this method, we obtain samples with RNA quality superior to the current standard in the LCM field, and show that low-input RNA-Seq kits that minimize PCR bias produce high fidelity sequencing metrics with less variability compared to current practices.
Figures
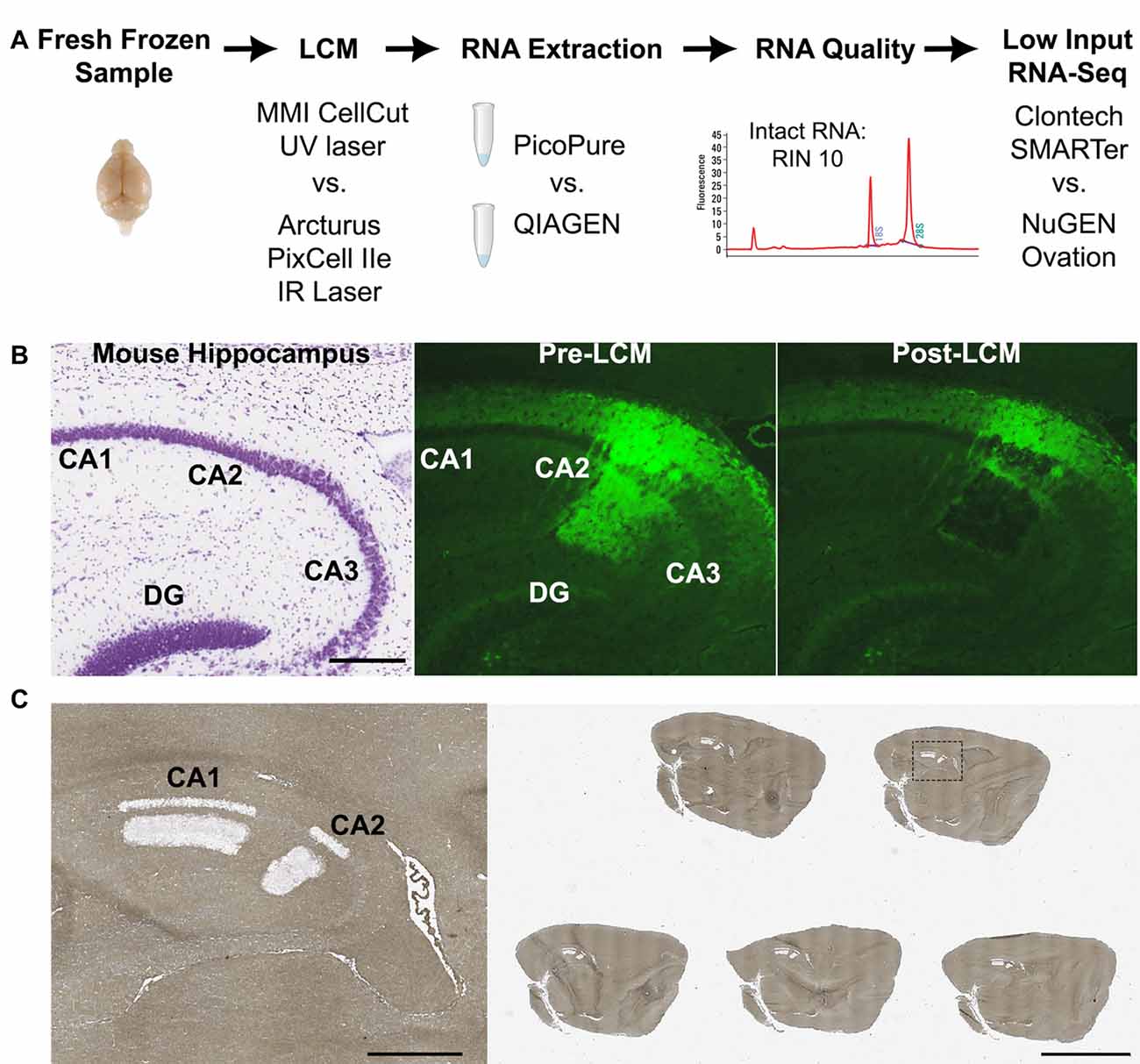
Figure 1. Laser capture-RNA-Seq workflow.
(A) Schematic of steps that were optimized in the current study (LCM instrument, RNA extraction and RNA-Seq library kit).
(B) Representative images of (left) nissl stained mouse hippocampus from Allen Brain Atlas, (middle) Amigo2-EGFP mouse hippocampus showing the fluorescently labeled CA2 cells and projections prior to LCM and (right) post LCM.
(C) Representative image of a glass slide with five dehydrated mouse brain sections post-LCM of hippocampal subregions CA1 and CA2. On the left is a magnified image of the dashed box.
Scale bars: 500 μm (B,C left) and 4 mm (C right).
CA, cornus ammonis; DG, dentate gyrus; LCM, laser capture microdissection; RIN, RNA Integrity Number.
- Figure 1 (139 KB)
{kind=link}
Figure 2. Comparison of RNA quality using different LCM methods.
(A) Graph comparing RNA quality (RIN) from LCM RNA samples captured using the MMI CellCut or Arcturus PixCell Instrument and extracted with either the Arcturus PicoPure Isolation kit or QIAGEN Micro RNeasy kit. An overall significant effect was found for both conditions using a two-way analyses of variance (ANOVA; CellCut vs. PixCell F(1,119) = 114.6; PicoPure vs. QIAGEN F(1,119) = 732.5). Although, it is important to note that two groups (Pixcell PicoPure and CellCut QIAGEN) were solely represented by one tissue type (see Experimental Summary in Table 1). There was also a significant interaction between the two conditions (Interaction F(1,119) = 9.177, p = 0.003).
(B) The same data shown in A plotted by tissue type. Each tissue (Hippocampus, Midbrain and Liver) showed a significant increase in RIN with the QIAGEN kits vs. PicoPure kits using Sidak’s multiple comparisons post hoc test. All data were normally distributed (passed KS normality test) and had similar variances as tested by Brown-Forsythe test.
(C,D) Representative Bioanalyzer gel (top) and electropherogram traces (bottom) from PixCell LCM RNA samples extracted using either the (C) Arcturus PicoPure Isolation kit or (D) QIAGEN Micro RNeasy kit. Note that these LCM samples were acquired simultaneously from different brain regions (CA1 vs. CA2) on the same sections from three mouse brains (#2, #4 or #6). Graphs are plotted min to max with a line at the mean. Numbers in parentheses indicate technical replicates.
####Overall group effect; ****post hoc result p < 0.0001; CB, cell body; DE, dendrite.
- Figure 2 (102 KB)
{kind=link}
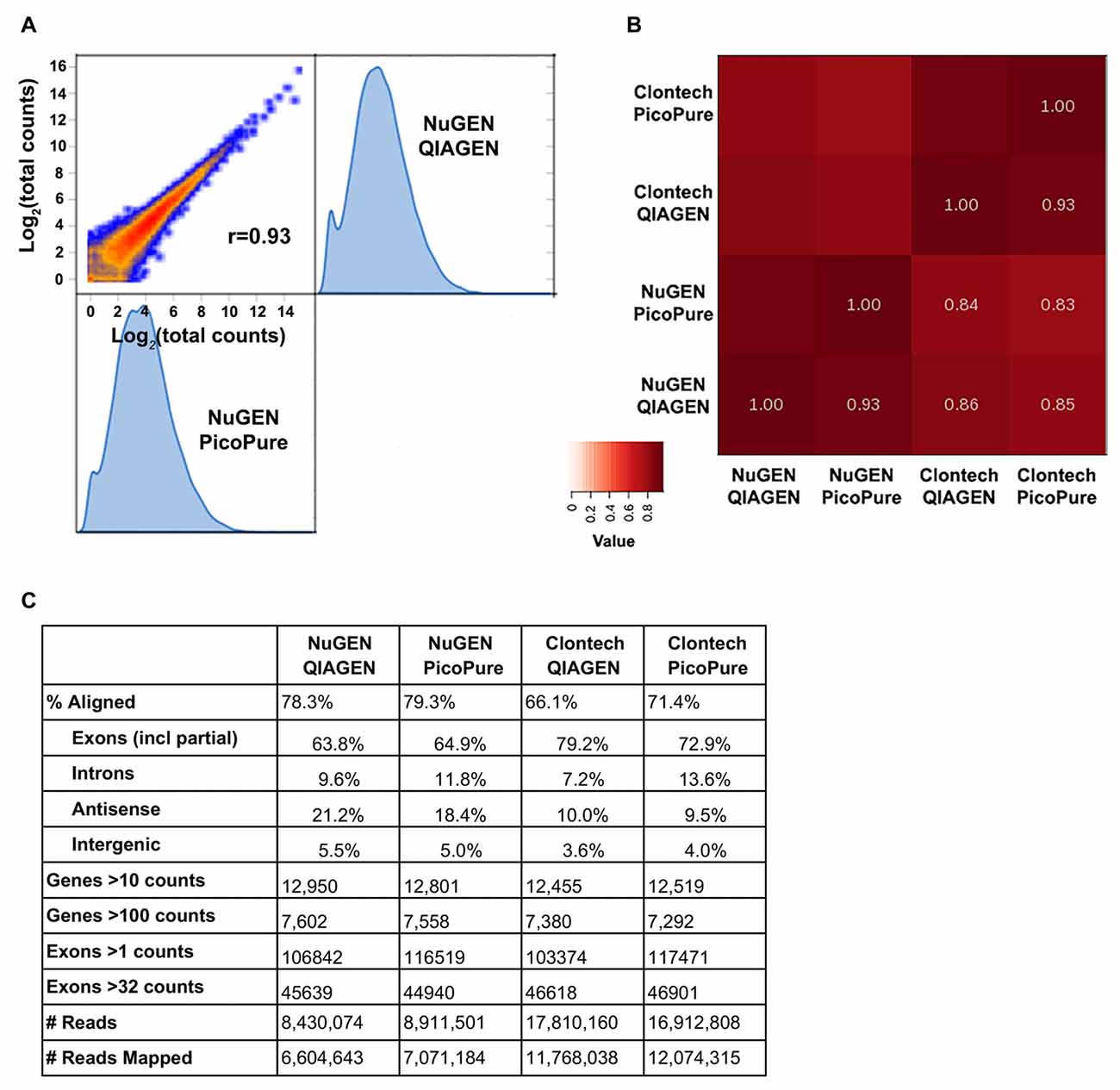
Figure 3. Sequencing metrics comparing library kits and RNA quality.
(A) Log2 expression correlation plot of NuGEN libraries made from high (QIAGEN) and low (PicoPure) quality RNA.
(B) Heat map summary of correlations from NuGEN and Clontech libraries. Note that the largest variation is from the library kit and not RNA quality.
(C) Table of sequencing metrics. Note that due to their larger library sizes, NuGEN samples obtained fewer total reads than Clontech.
Data were normalized for read depth prior to gene and exon analyses.
- Figure 3 (146 KB)
{kind=link}
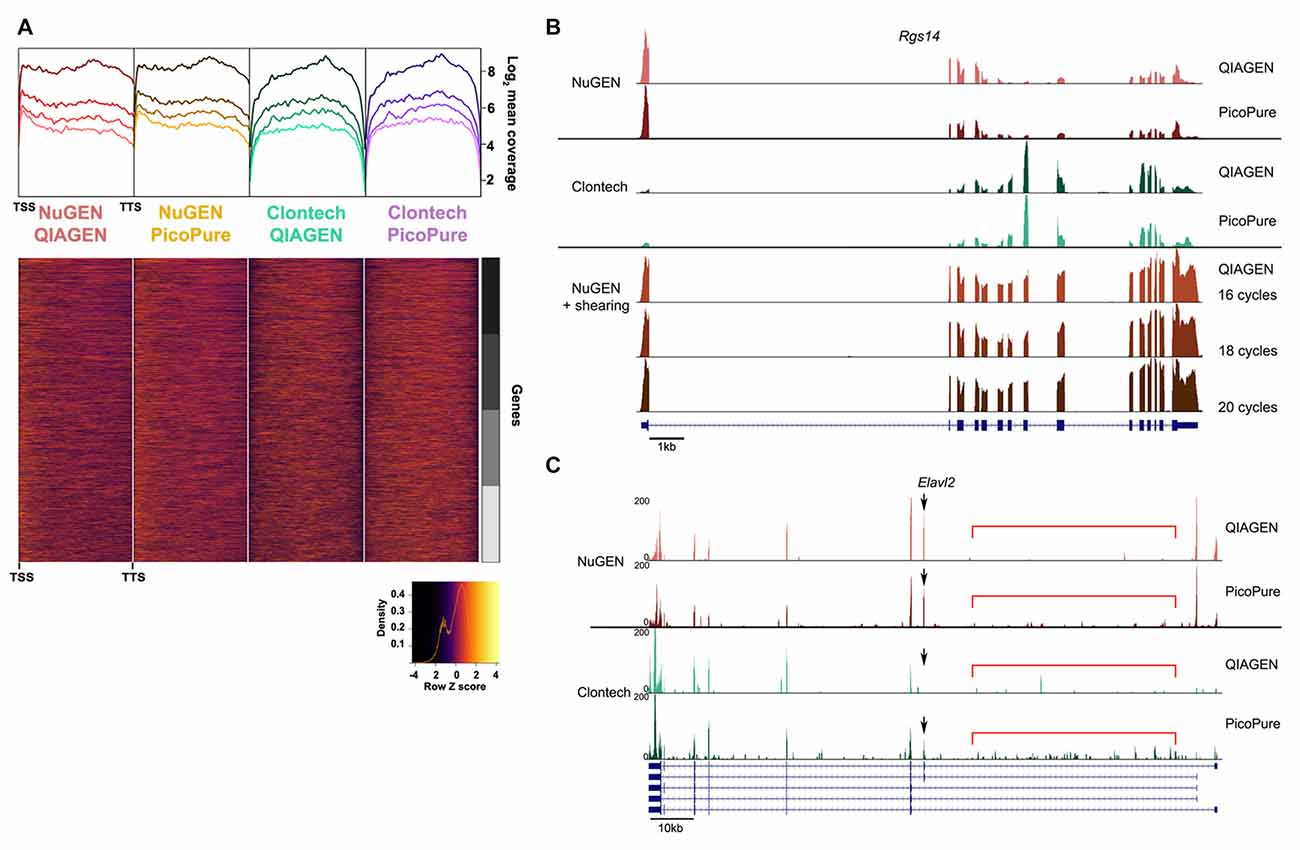
Figure 4. Comparison of gene coverage.
(A) 5′-3′ average gene coverage for each sample. (Top) Gene coverage is broken down by expression with the darker colors corresponding to the highest expressing genes. (Bottom) Scaled heat map of the gene coverage graphed above. Note that libraries made with the NuGEN kit have greater gene coverage at the 5′ and 3′ ends compared to libraries made with the Clontech kit. (B,C) Representative gene loci to visualize read coverage from each sample.
(B) Rgs14 locus illustrates the greater 5′ and 3′ end coverage in the NuGEN samples. The 5′ bias in the NuGEN samples is remedied by shearing. NuGEN gene coverage is unaltered by increasing cycles of PCR.
(C) Elavl2 locus illustrates the increased intronic read coverage (denoted by red bar) seen in PicoPure or low quality samples. Differential isoform detection is denoted with a black arrow.
- Figure 4 (105 KB)
{kind=link}
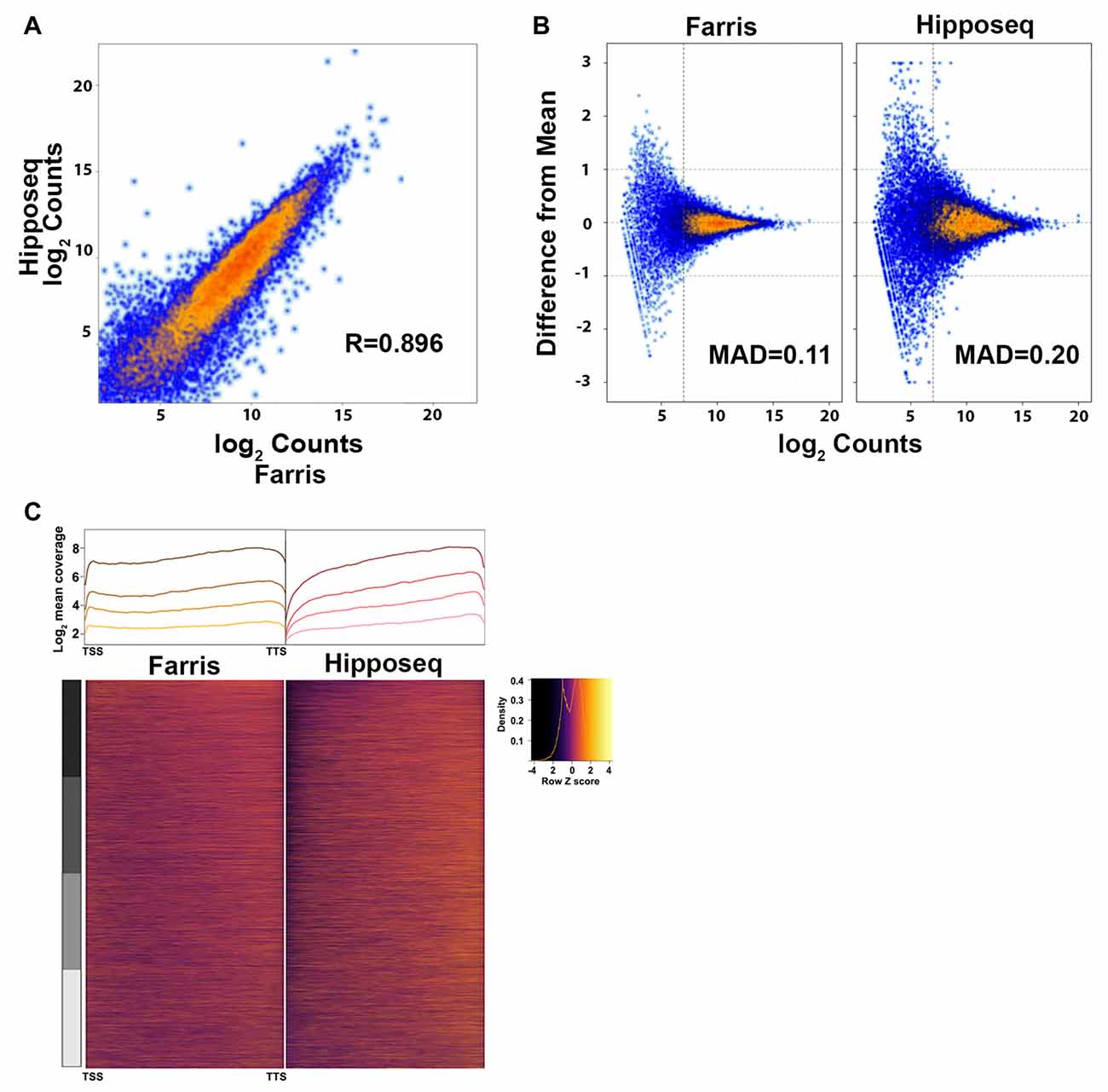
Figure 5. Cross-correlation of RNA-Seq expression data.
(A) Correlation plot of the CA2 RNA-Seq mean expression data presented here (N = 3) vs. the CA2 RNA-Seq data published in Cembrowski et al. (2016) referred to here as Hipposeq (http://hipposeq.janelia.org, N = 3).
(B) MA plots of a representative sample from each dataset depicting the mean absolute deviation (MAD) vs. abundance. The threshold for noise is drawn as a vertical line.
(C) 5′-3′ average gene coverage for each dataset. (Top) Gene coverage is broken down by expression with the darker colors corresponding to the highest expressing genes. (Bottom) Scaled heat map of the gene coverage graphed above.
- Figure 5 (118 KB)
{kind=link}
Tables
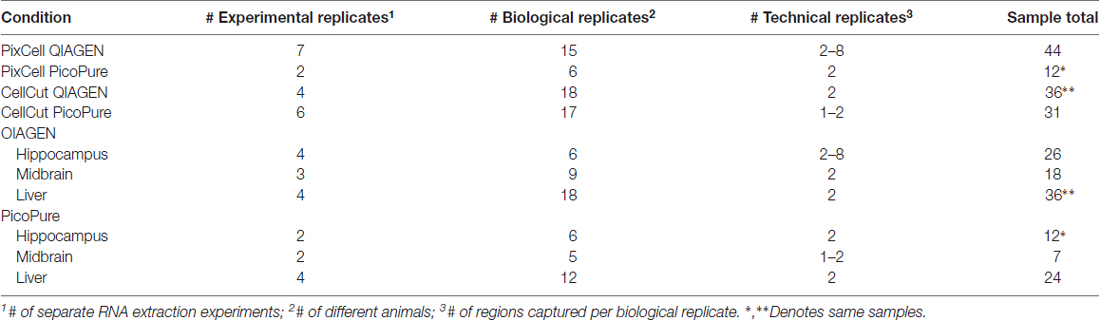
Table 1. Experimental summary of laser capture microdissection (LCM) samples.
- Table 1 (145 KB)
{kind=link}
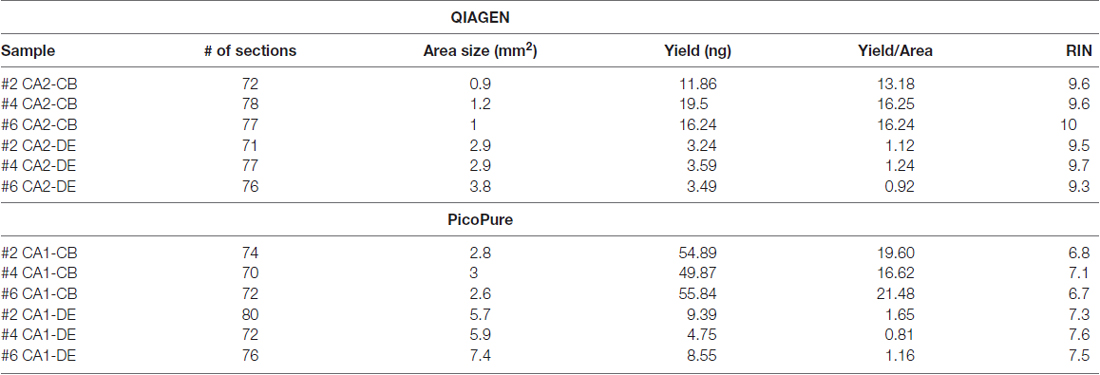
Table 2. Within sample comparison of RNA quality from PixCell laser captured tissues.
- Table 2 (162 KB)
{kind=link}