

ORIO (Online Resource for Integrative Omics): A Web-Based Platform for Rapid Integration of Next Generation Sequencing Data
Lavender CA, Shapiro AJ, Burkholder AB, Bennett BD, Adelman K, Fargo DC.
Nucleic Acids Research (2017)
DOI: https://doi.org/10.1093/nar/gkx270
PMID: 27933809
Publication
Abstract
Established and emerging next generation sequencing (NGS)-based technologies allow for genome-wide interrogation of diverse biological processes. However, accessibility of NGS data remains a problem, and few user-friendly resources exist for integrative analysis of NGS data from different sources and experimental techniques. Here, we present Online Resource for Integrative Omics (ORIO; https://orio.niehs.nih.gov/), a web-based resource with an intuitive user interface for rapid analysis and integration of NGS data. To use ORIO, the user specifies NGS data of interest along with a list of genomic coordinates. Genomic coordinates may be biologically relevant features from a variety of sources, such as ChIP-seq peaks for a given protein or transcription start sites from known gene models. ORIO first iteratively finds read coverage values at each genomic feature for each NGS dataset. Data are then integrated using clustering-based approaches, giving hierarchical relationships across NGS datasets and separating individual genomic features into groups. In focusing its analysis on read coverage, ORIO makes limited assumptions about the analyzed data; this allows the tool to be applied across data from a variety of experiments and techniques. Results from analysis are presented in dynamic displays alongside user-controlled statistical tests, supporting rapid statistical validation of observed results. We emphasize the versatility of ORIO through diverse examples, ranging from NGS data quality control to characterization of enhancer regions and integration of gene expression information. Easily accessible on a public web server, we anticipate wide use of ORIO in genome-wide investigations by life scientists.
Figures
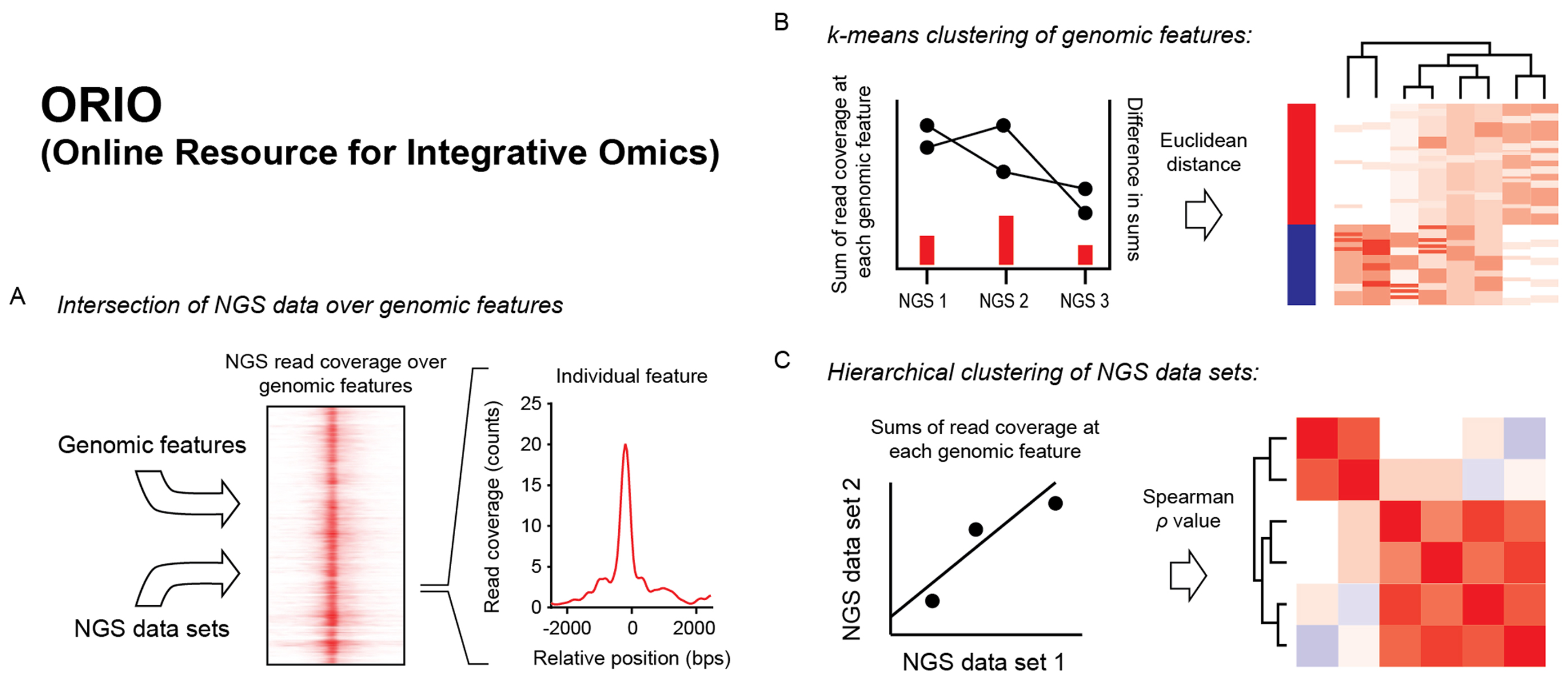
Figure 1. Schematic of analysis by ORIO.
(A) Intersection of NGS data over genomic features. ORIO first finds read coverage values at each genomic feature for each NGS dataset in an analysis. Read coverage value are determined for genomic windows anchored on feature positions.
(B) k-means clustering of genomic features. Read coverage values are used to cluster genomic features by k-means. To perform clustering, read coverage values for each NGS dataset are concatenated to make a 1D vector for each feature. The Euclidean distances between these vectors are then used in k-means clustering.
(C) Hierarchical clustering of NGS datasets. Pairwise correlation values considering read coverage values at each feature are used as a distance metric in hierarchical clustering of datasets.
- Figure 1 (696 KB)
{kind=link}
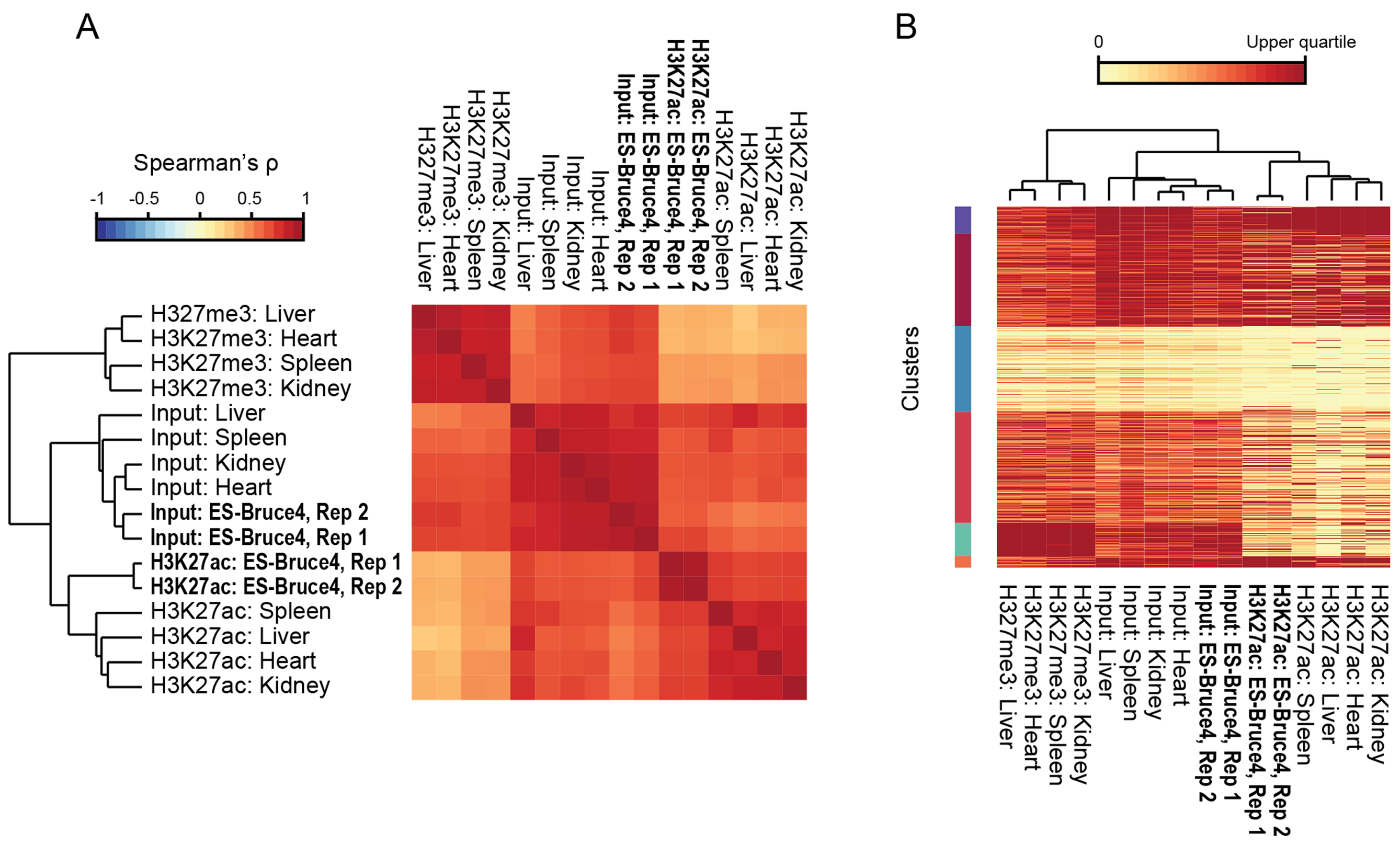
Figure 2. Example 1: Validation of test H3K27ac ChIP-seq data.
(A) NGS dataset clustering. ORIO analysis was performed for ChIP-seq experiments from diverse tissues considering mouse RefSeq transcription start sites. H3K27ac datasets were run with input controls and the mutually exclusive H3K27me3 mark. Hierarchical clustering of samples is shown by dendrogram.
(B) Clustering of transcription start sites by ChIP-seq data. Read coverage at transcription start sites is presented by heatmap where rows correspond to individual features and columns correspond to ChIP-seq datasets. Clusters (k-means; k = 6) are given on the left side of the plot. The dendrogram (top) reflects hierarchical clustering of ChIP-seq datasets shown in A. All plots were generated using ORIO.
- Figure 2 (1 MB)
{kind=link}
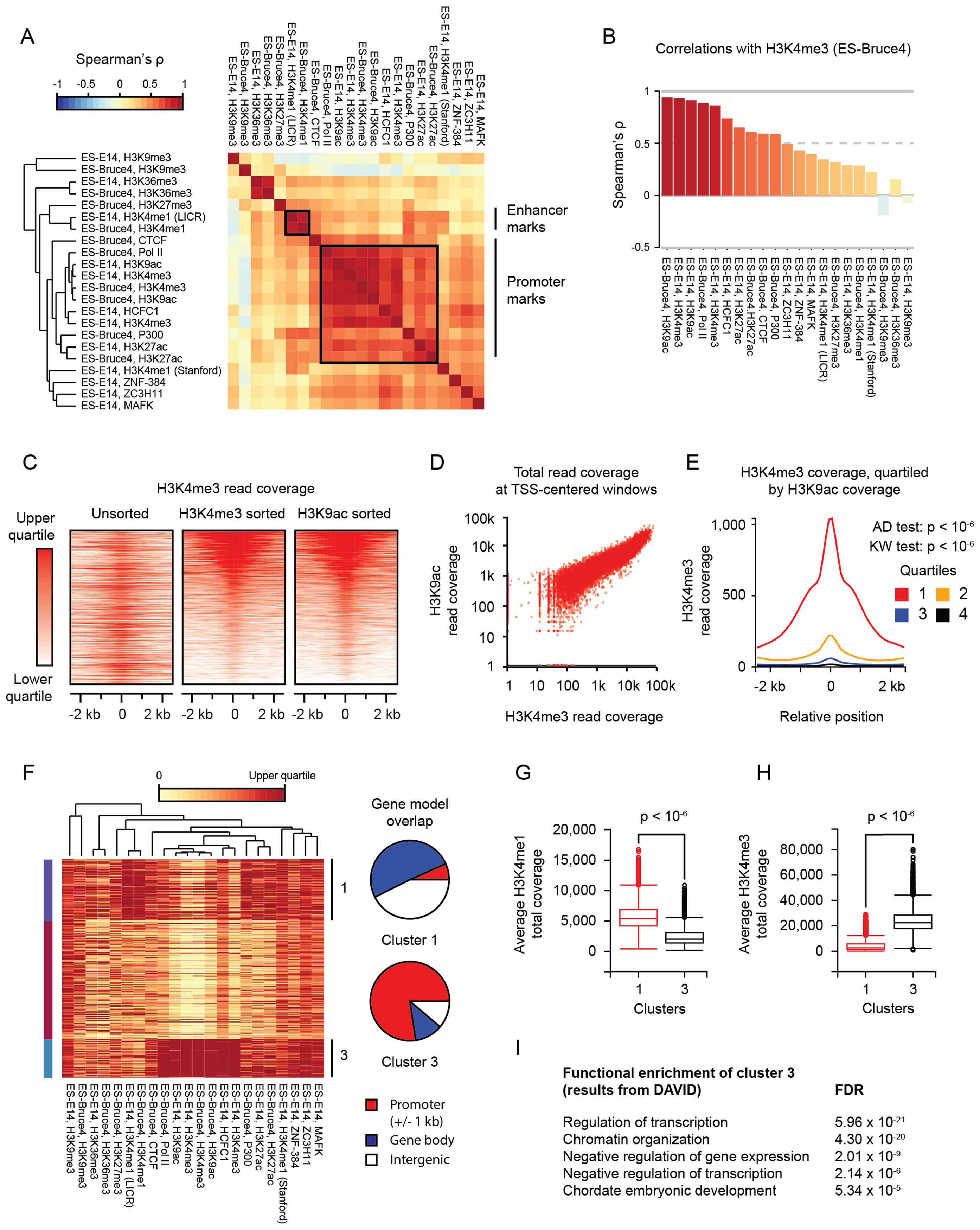
Figure 3. Example 2: ORIO analysis of H3K27ac ChIP-seq peaks.
(A) NGS dataset clustering. ORIO analysis was performed for ES-Bruce4 and ES-E14 ChIP-seq experiments from ENCODE considering mouse ES-Bruce4 H3K27ac ChIP-seq peaks. Pairwise Spearman correlation values are displayed by heatmap with hierarchical clustering of datasets indicated by the inset dendrogram. Groups of enhancer- and promoter-associated are indicated by boxes and by labels along the right side.
(B) Boxplot of pairwise correlations (Spearman's ρ) with H3K4me3 ChIP-seq data.
(C) Rank-ordered heatmaps of H3K4me3 read coverage over H3K27ac peaks. Heatmaps show read coverage from H3K4me3 ChIP-seq in ES-Bruce4 cells. From left to right, heatmap rows are unsorted, sorted by decreasing totals of H3K4me3 coverage, and sorted by decreasing totals of H3K9ac coverage.
(D) Scatterplot ofH3K4me3 and H3K9ac read coverage over windows centered on H3K27ac peaks.
(E) Plots of average H3K4me3 read coverage across genomic windows centered on H3K27ac peaks. Average coverages are quartiled by H3K9ac read coverage: the first quartile has the highest H3K9ac read coverage, and the fourth quartile has the lowest. P-values are found for Anderson–Darling (AD) and Kruskal–Wallis (KW) tests considering quartiled distributions of H3K4me3 read coverage.
(F) Clustering of H3K27ac peaks as genomic features. Read coverage is displayed by heatmap, with each row corresponding to an individual feature and each column corresponding to an NGS dataset. NGS clustering is shown by dendrogram. Clusters (k-means; k = 3) are indicated along the left and right sides. Overlap of cluster features with RefSeq gene models is shown by pie charts.
(G and H) Box plots of H3K4me1 and H3K4me3 read coverage in feature clusters 1 and 3. P-values were determined by Mann–Whitney test.
(I) Gene ontology results for genes nearest H3K7ac peaks in cluster 3. Gene ontology analysis was performed by DAVID (29).
All plots were generated using ORIO, with the except of pie charts in F; pie charts were generated using matplotlib.
- Figure 3 (3 MB)
{kind=link}
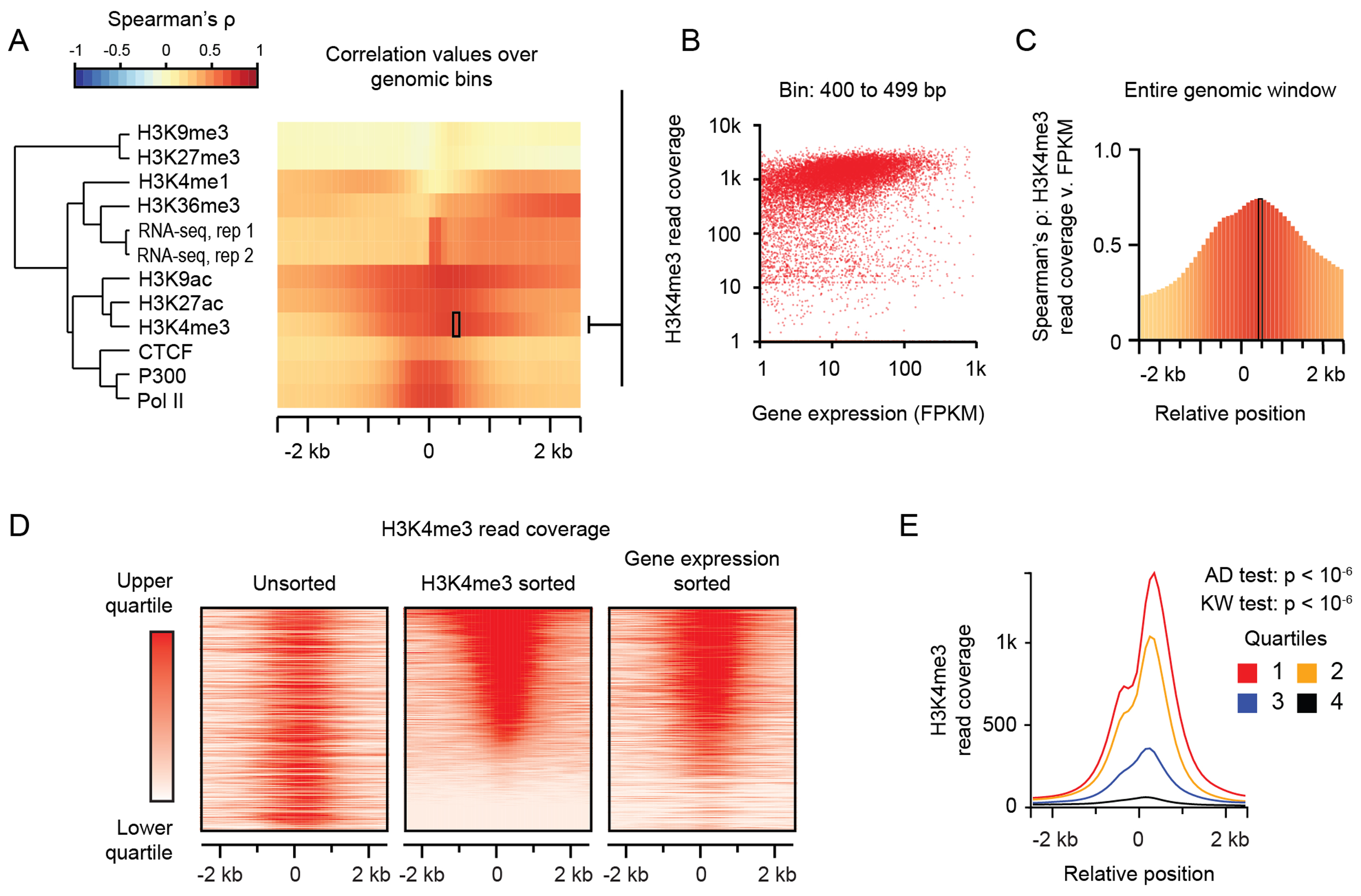
Figure 4. Example 3: ORIO analysis integrating gene expression with ChIP-seq and RNA-seq.
Example 3: ORIO analysis integrating gene expression with ChIP-seq and RNA-seq read coverage information. (A) Heatmap of correlations of read coverage values at transcription start sites with gene expression (FPKM). Each row corresponds to an individual NGS dataset, and each column corresponds to a genomic window bin. The left-side dendrogram was generated by hierarchical clustering of correlation values. (B) Scatterplot of gene expression values and H3K4me3 read coverage across transcription start sites in the 400–499 bp bin. This bin is indicated by the box in A. (C) Plot of correlation values across all window bins. Spearman correlation values were computed considering H3K4me3 read coverage at a RefSeq TSS and gene expression values (FPKM) at the nearest gene model. (D) Rank-ordered heatmaps of H3K4me3 read coverage. From left to right, heatmaps of H3K4me3 read coverage are unsorted, sorted by decreasing H3K4me3 read coverage, and sorted by decreasing gene expression of the closest gene as measured by FPKM. (E) Plots of average H3K4me3 read coverage for transcription start sites quartiled by gene expression. The first quartile corresponds to genes with the highest expression. Listed P-values are from Anderson–Darling (AD) and Kruskal–Wallis (KW) tests considering the quartiled distributions of total H3K4me3 read coverage. Plots were generated using ORIO.
- Figure 4 (1 MB)
{kind=link}
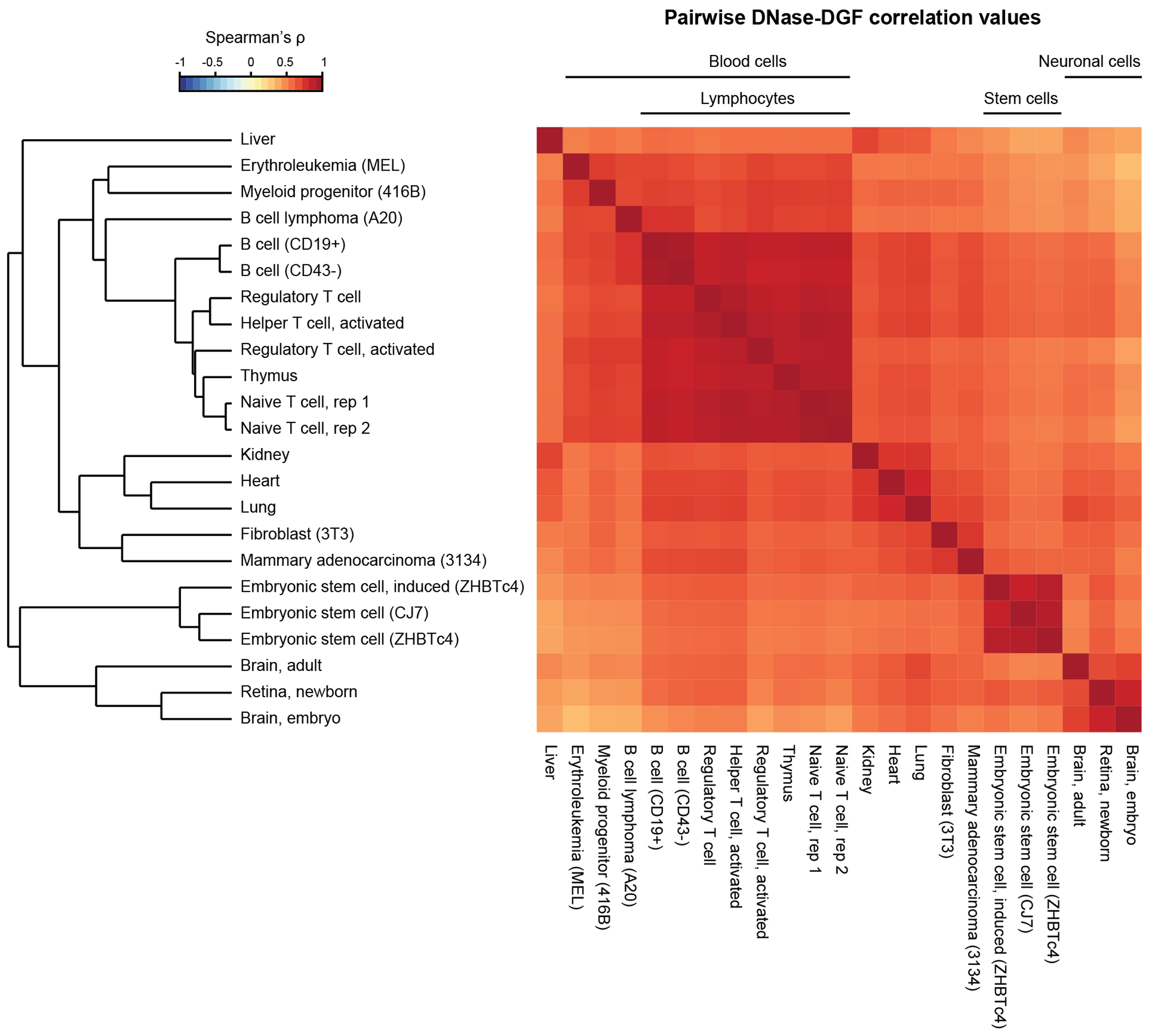
Figure 5. Example 4: Clustering of mouse DNase-DGF datasets by ORIO analysis.
ENCODE DNase-DGF datasets from 22 cell and tissue types (6,36) were analyzed considering a feature list of 163,274 mouse TSSs. Correlation values (Spearman ρ) were found for each dataset pair and are displayed on a heatmap. Hierarchical clustering results were derived based on these correlation values and accurate recapitulate cell lineages. Clustering results are presented as a dendrogram, and cell subgroups are indicated along the top of the heatmap. Plots were generated using ORIO.
- Figure 5 (1 MB)
{kind=link}
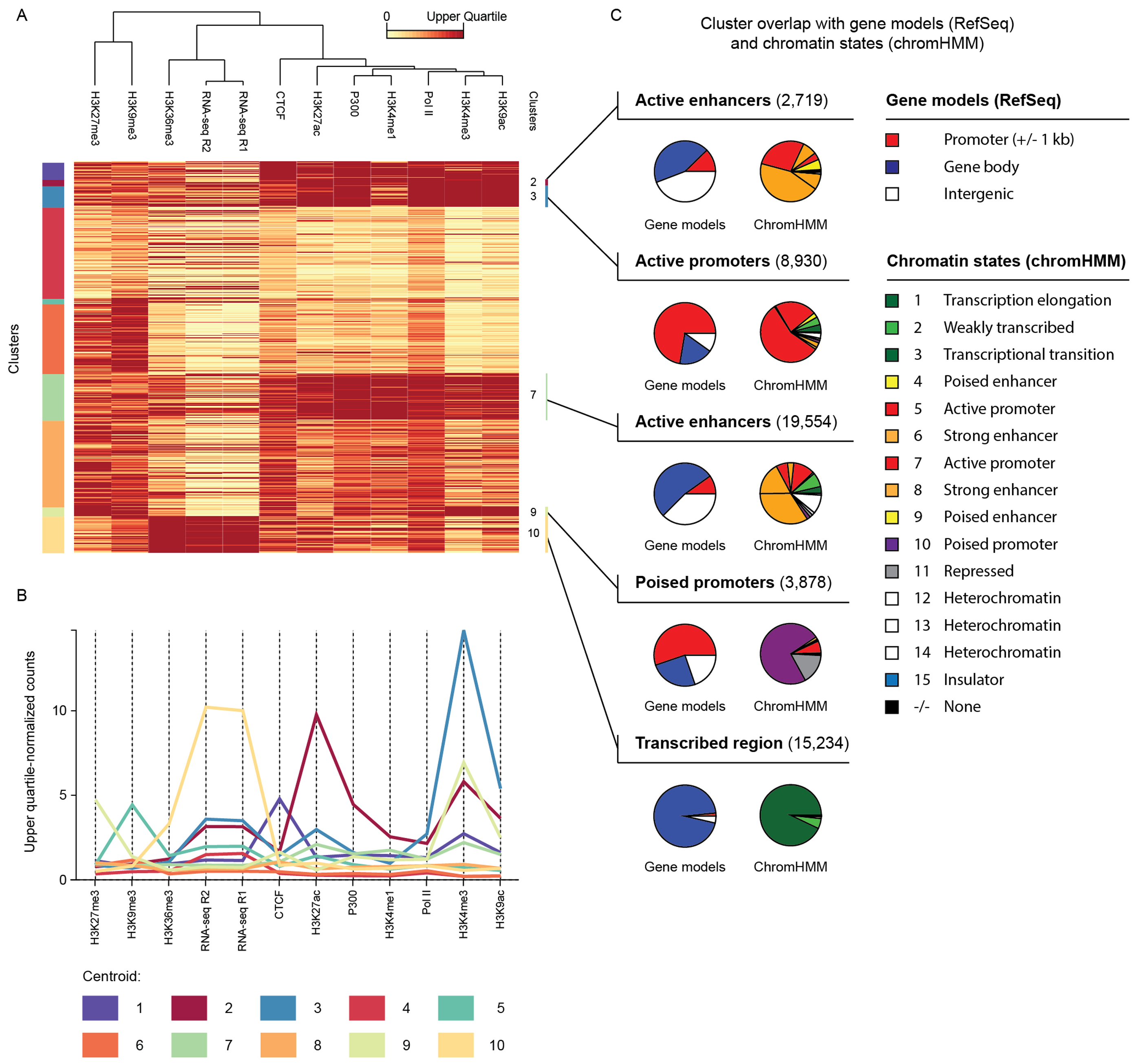
Figure 6. Example 5: ORIO analysis of mouse embryonic stem cell TSSs.
(A) Clusters (k-means; k = 10) of TSSs called from embryonic stem cell Start-seq data. Clustering was performed considering observing read coverage of ENCODE Bruce4 C57BL/6 mouse embryonic stem cell datasets (6). Clustering results are displayed on an upper quartile-normalized heatmap where rows correspond to TSSs and columns to NGS datasets. Cluster membership is given by vertical bars on the left edge of the heatmap. (B) Normalized coverage values for the centroid of each cluster. Each column corresponds to an analyzed dataset. (C) Overlap of each cluster with RefSeq gene models (17) and chromHMM chromatin states (38,39). Overlap is displayed using pie charts. The redundancy of chromHMM state names is characteristic of this approach with labels such as ‘strong enhancer’ and ‘heterochromatin’ being applied to multiple states. Clustering heatmap and centroid plot were generated using ORIO; pie charts were generated using matplotlib (43).
- Figure 6 (2 MB)
{kind=link}
Tables
Table 1. Benchmarking of ORIO runtimes with analyses of different sizes.
- Table 1 (84 KB)
Table 2. Comparison of ORIO features with similar tools.
- Table 2 (211 KB)